Citation Information
Shankarappa R, Margolick JB, Gange SJ, Rodrigo AG, Upchurch D, Farzadegan H, Gupta P, Rinaldo CR, Learn GH, He X, Huang XL, Mullins JI (1999). Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. Journal of virology, 73(12), 10489-502. (pubmed)
Abstract
To understand the high variability of the asymptomatic interval between primary human immunodeficiency virus type 1 (HIV-1) infection and the development of AIDS, we studied the evolution of the C2-V5 region of the HIV-1 env gene and of T-cell subsets in nine men with a moderate or slow rate of disease progression. They were monitored from the time of seroconversion for a period of 6 to 12 years until the development of advanced disease in seven men. Based on the analysis of viral divergence from the founder strain, viral population diversity within sequential time points, and the outgrowth of viruses capable of utilizing the CXCR4 receptor (X4 viruses), the existence of three distinct phases within the asymptomatic interval is suggested: an early phase of variable duration during which linear increases ( approximately 1% per year) in both divergence and diversity were observed; an intermediate phase lasting an average of 1.8 years, characterized by a continued increase in divergence but with stabilization or decline in diversity; and a late phase characterized by a slowdown or stabilization of divergence and continued stability or decline in diversity. X4 variants emerged around the time of the early- to intermediate-phase transition and then achieved peak representation and began a decline around the transition between the intermediate and late phases. The late-phase transition was also associated with failure of T-cell homeostasis (defined by a downward inflection in CD3(+) T cells) and decline of CD4(+) T cells to </=200 cells/microliter. The strength of these temporal associations between viral divergence and diversity, viral coreceptor specificity, and T-cell homeostasis and subset composition supports the concept that the phases described represent a consistent pattern of viral evolution during the course of HIV-1 infection in moderate progressors. Recognition of this pattern may help explain previous conflicting data on the relationship between viral evolution and disease progression and may provide a useful framework for evaluating immune damage and recovery in untreated and treated HIV-1 infections.
Supplemental Data
Correction
The equation in the sentence:
Under a neutral model of evolution, \(s_{a_0}\) is one randomly selected sequence, and \( E[d(s_{a_0}, s_1)] = E(d(s_{0_x}, s_{0_y})] \) where the right-hand term…
(p. 10490, right column, second paragraph)
should be:
\( E[d(s_{a_0}, s_0)] = E[d(s_{0_x}, s_{0_y})] \)
We are grateful to Anders Kvist of Lund University for bringing this error to our attention.
Sequences
Genbank Accession numbers AF137629-AF138163, AF138166-AF138263, and AF138305-AF138703 correspond to sequences for this study. Each sequence record contains the subject ID, whether the sequence was sampled from peripheral blood mononuclear cells (PBMC) or from plasma, and time following seroconversion. This is illustrated below for two sequences:
LOCUS AF137716 597 bp RNA linear VRL 08-FEB-2000
DEFINITION HIV-1 p1p080-326 from USA envelope glycoprotein (env) gene, partial
cds.
LOCUS AF137715 597 bp DNA linear VRL 08-FEB-2000
DEFINITION HIV-1 p1c077-317 from USA envelope glycoprotein (env) gene, partial
cds.Both of these are from subject #1 (p1). AF137716, clone number 326, was sampled from plasma (p for plasma) collected 80 months post seroconversion. Similarly, the AF137715, clone number 317, was from PBMC (c for cellular) sampled 77 months post seroconversion.
Nucleotide alignments from this study are available in fasta or nexus format.
Appendix Materials

Appendix Figure 1(gif pdf) Phylogenetic characterization of 1300 sequences sampled from the 9 participants described in this study and six selected sequences from GenBank. A neighbor-joining tree was constructed using maximum-likelihood distances in Phylip v3.5 (1) after regions that could not be unambiguously aligned were removed. Sequences sampled from each study participant formed monophyletic clusters separate from the other participants as well as from the six unrelated sequences included in this illustration. No viral sequence from this study was found to be closely related to prototypic HIV-1 sequences or to other known strains present or sequences determined in the laboratory. These observations are consistent with an absence of sample contamination or mix-up (3).
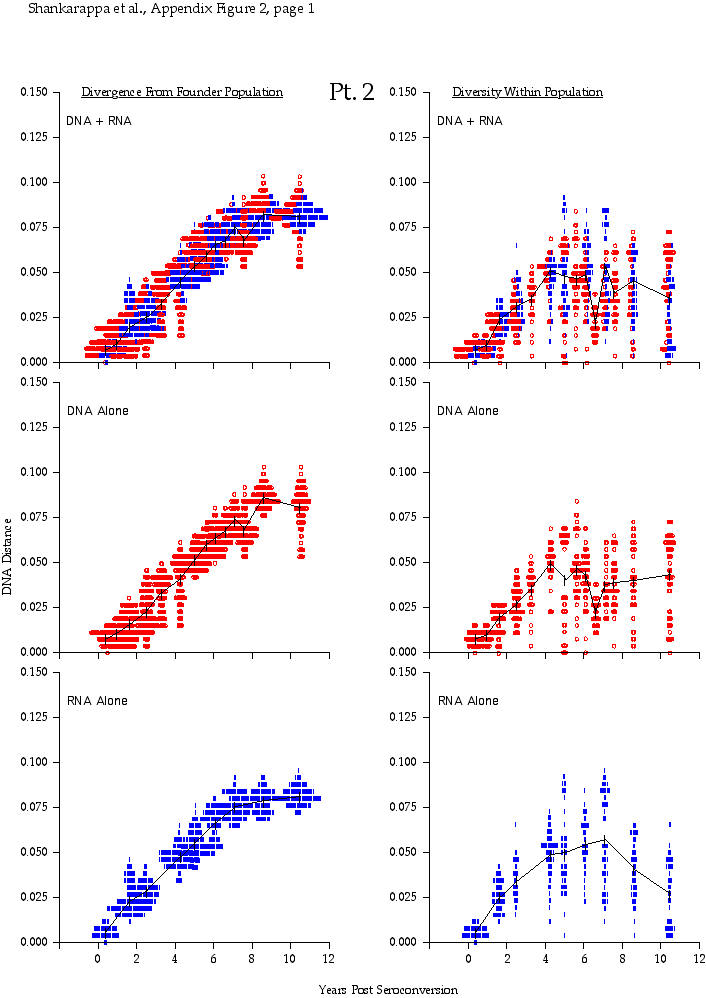
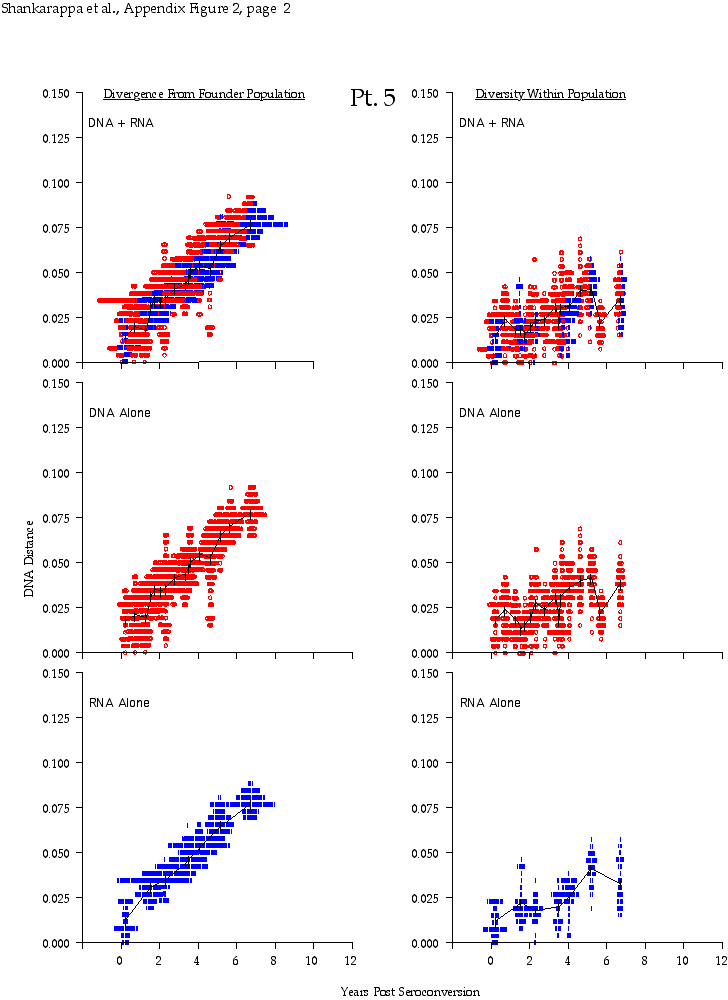
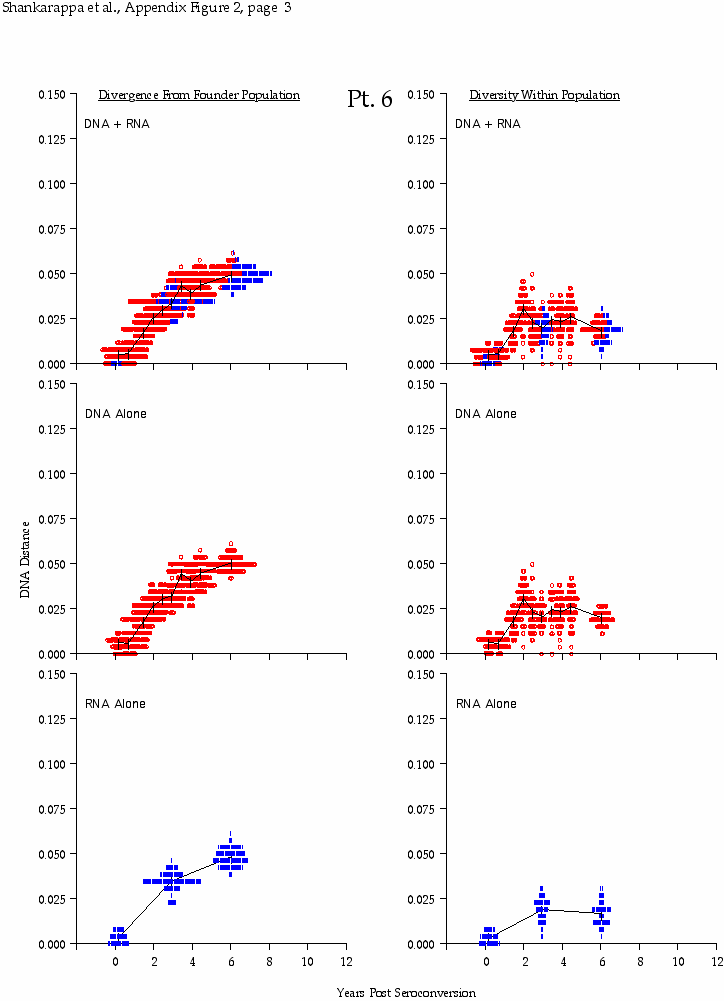
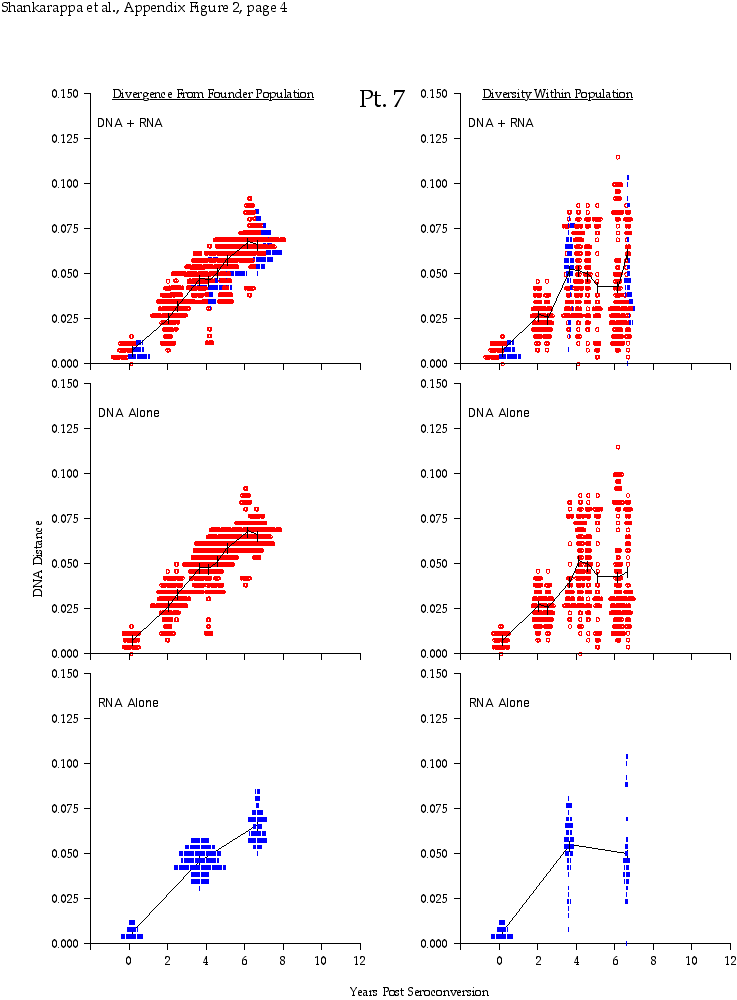
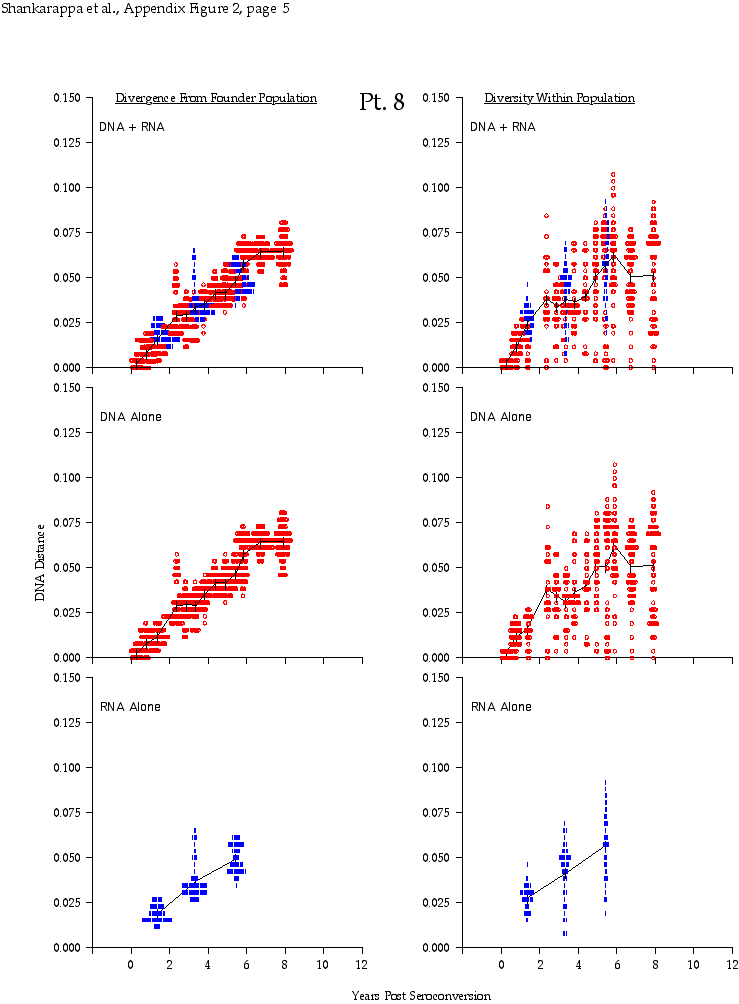
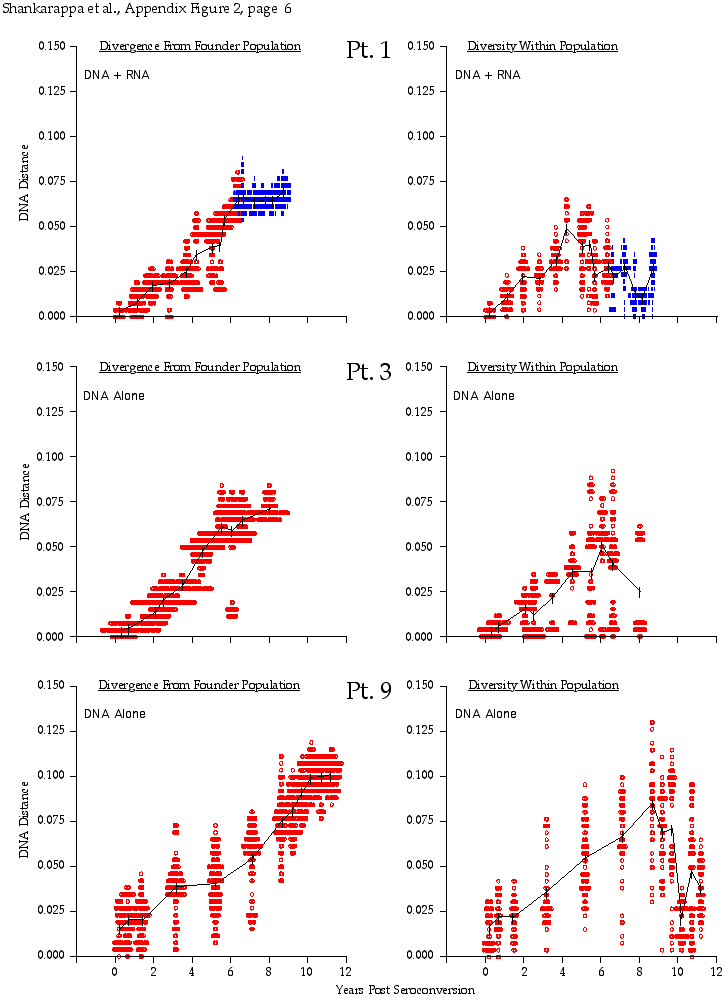
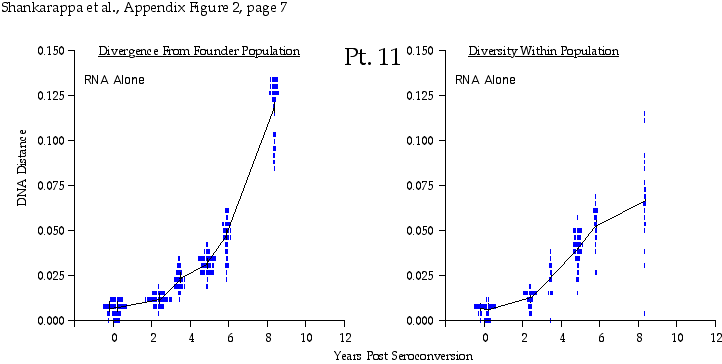
Appendix Figure 2 (pdf, single-page gif: 1 2 3 4 5 6 7) Pairwise comparisons of virus population divergence from the founder population (panels on the left side of the figure) and diversity within each sample (panels on the right) in each of the nine participants. Pairwise distances were estimated using the Kimura 2-parameter model of viral evolution (2). These distances were plotted on the y-axis at the corresponding times following seroconversion. The lines within each panel connect the mean values for each time point. Comparisons of sequences from PBMC are shown with a red open circle for each data point, those from plasma are shown with a blue vertical line for each data point. The first 5 panels show the estimates of viral diversification for participants in which sequences were sampled from both PBMC and plasma. Subsequent panels show these estimates sequences sampled from PBMC DNA or from plasma RNA (but not both at any time point).
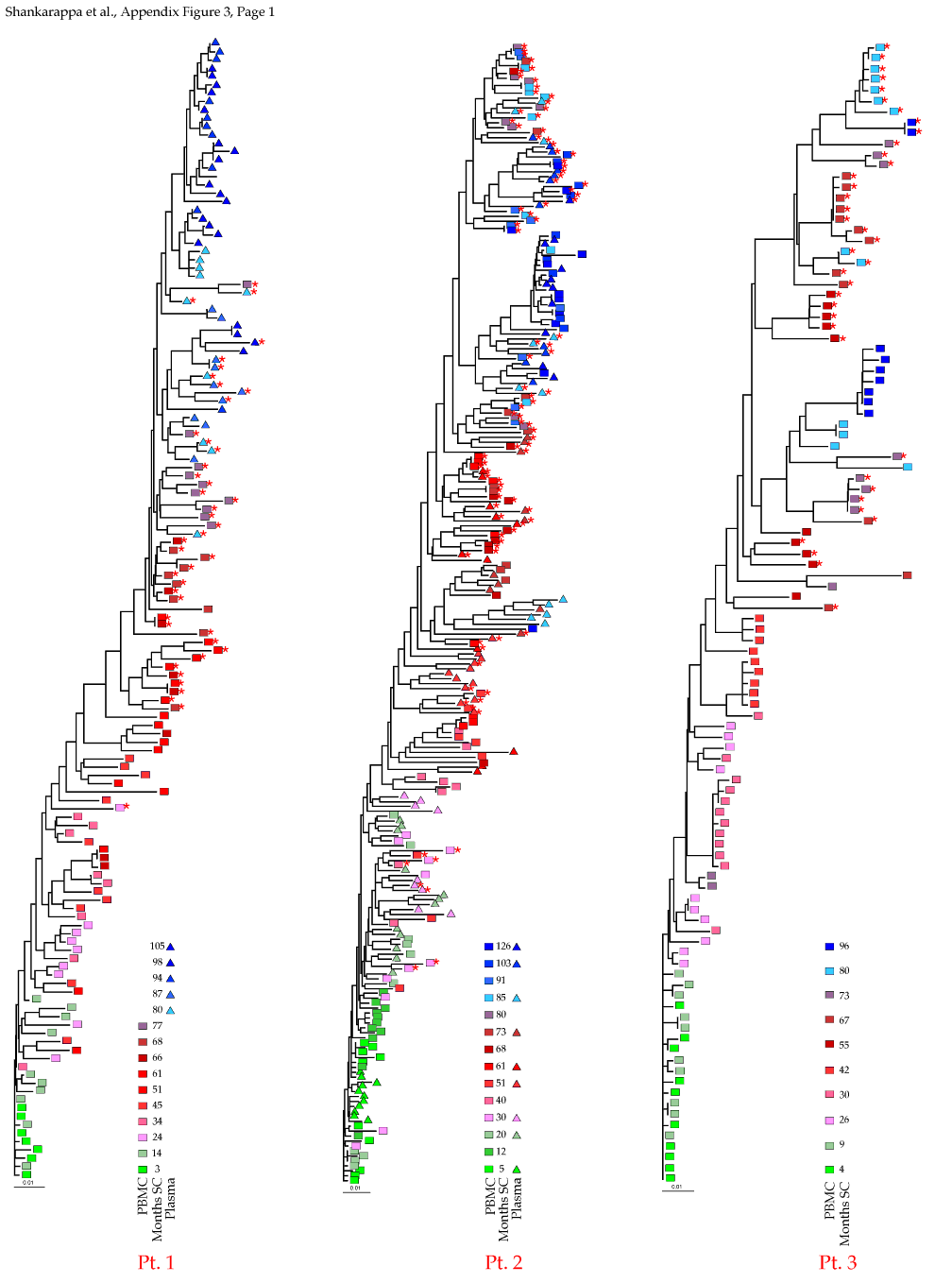
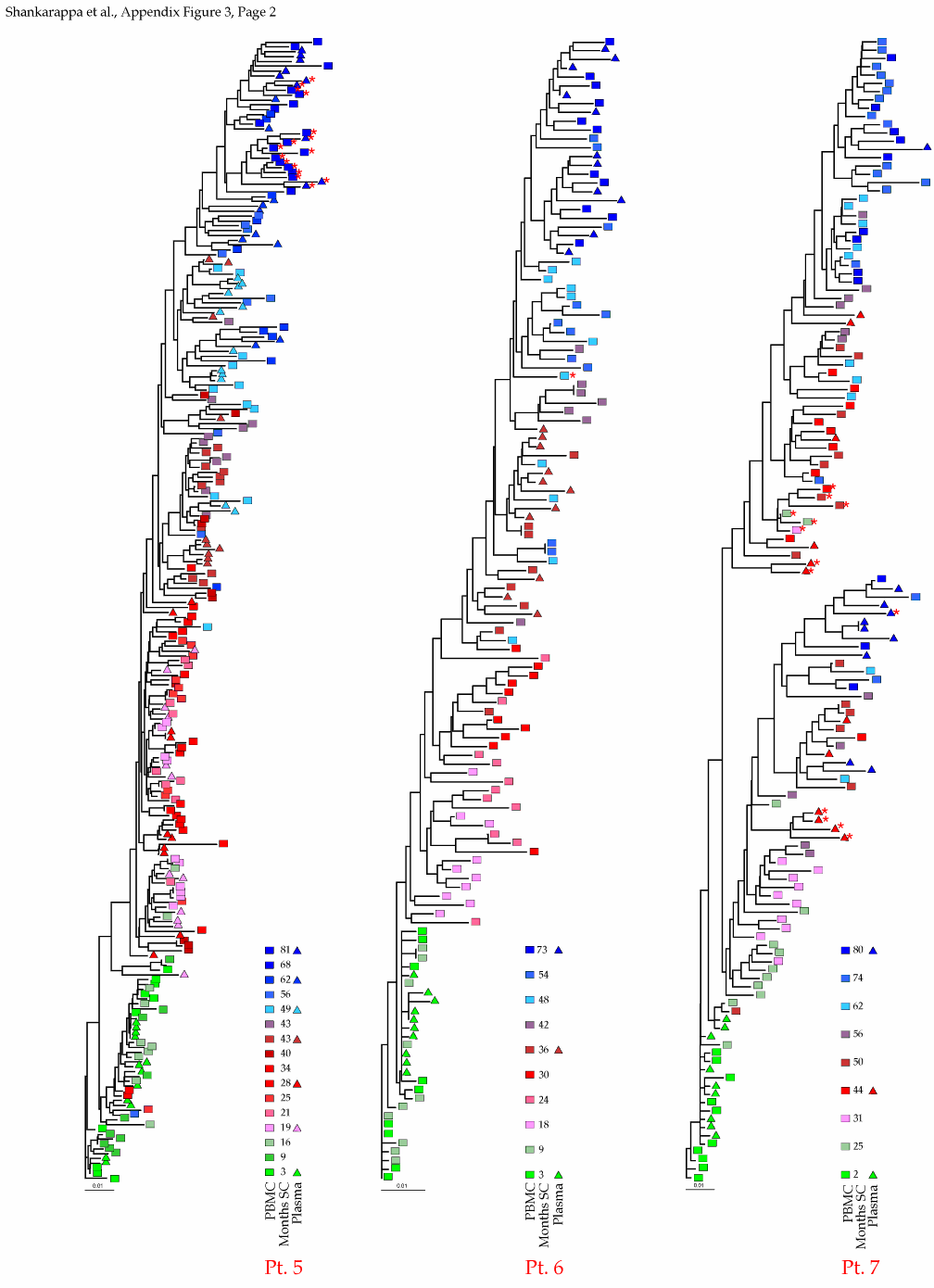
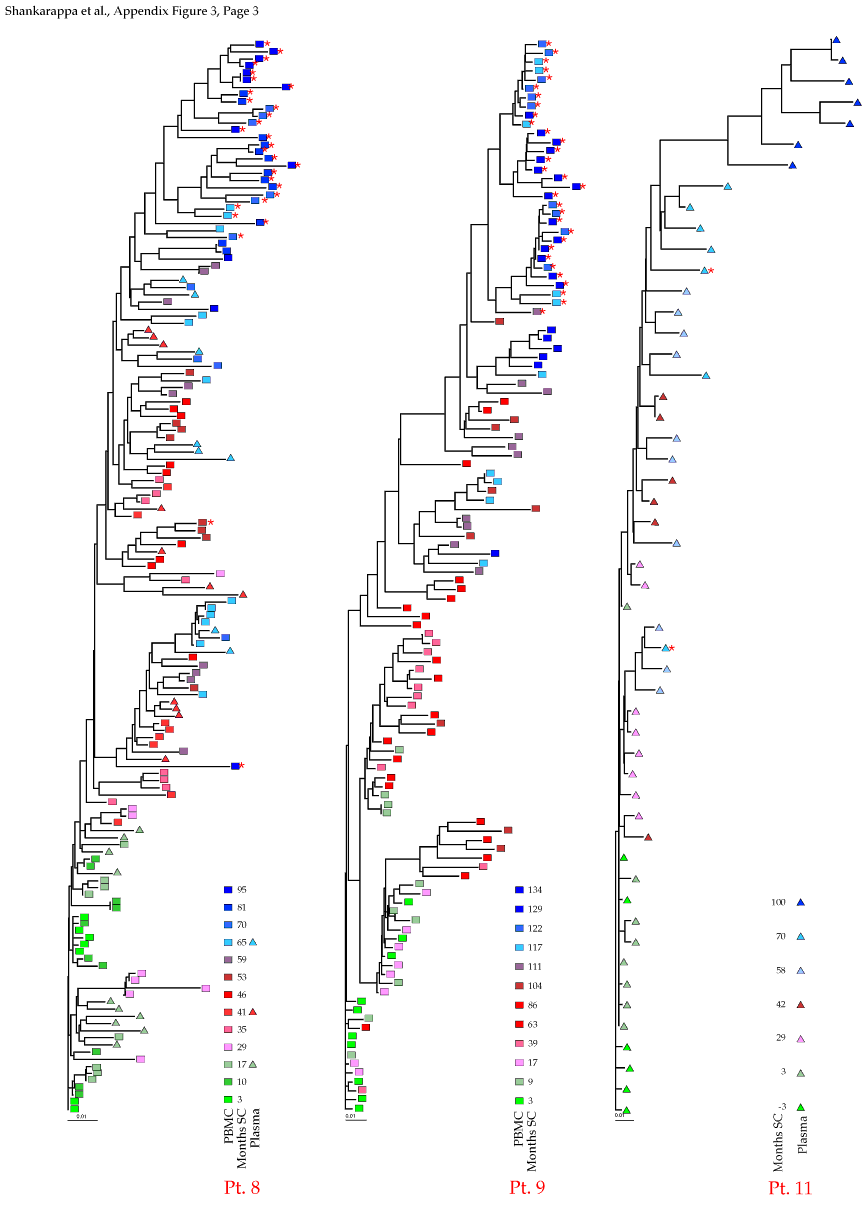
Appendix Figure 3 (pdf, single-page gif: 1 2 3) Phylogenetic analysis of HIV-1 env C2-V5 sequences from each of the nine participants. Sequences were aligned and visually adjusted using GDE (4). The phylograms were estimated by the method of neighbor-joining from a matrix of pairwise maximum likelihood sequence distances (transition/transversion ratio = 1.4) using programs from version 3.5 of the PHYLIP package (1). Trees were rooted to one of the sequences sampled at the first time point using Treeview (from R. Page). The legend for each phylogram shows the time of sampling in months following seroconversion and is depicted in an arbitrary color gradient. DNA sequences from PBMC are depicted as squares and plasma RNA sequences as triangles. Sequence changes expected to confer an X4 phenotype (SI phenotype on MT-2 cells and CXCR4 co-receptor specificity) based on basic amino acid substitution at any of the three positions (#306, 319, 320 of gp120) are indicated by a red asterisk.
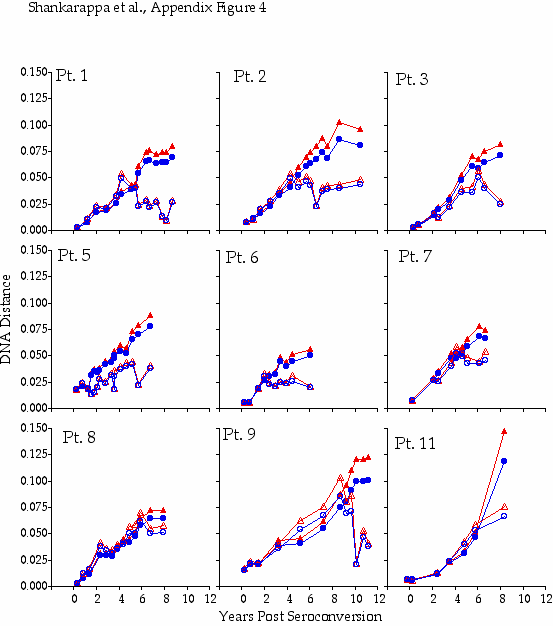
Appendix Figure 4 (pdf, gif) Comparison of DNA distance estimations in nine participants using Kimura 2-parameter (K2P) model of evolution with a transition to transversion ratio of 2 (2) vs. a General Time Reversible (GTR) model with a site-to-site variation in substitution rates (discrete approximation of a γ distribution with a shape parameter,α=0.5) (5). DNA distance estimates are shown for sequences from PBMC only in all participants except for Participant1, where plasma data is shown for the time points when PBMC samples were not available, and Participant 11 for whom insufficient amounts of viral DNA was found in PBMC. K2P distances are plotted as blue circles and GTR DNA distances are plotted as red triangles. Intra-time point DNA diversity is depicted by open symbols and the DNA distance as compared to founder sequences is shown by filled symbols. These comparisons show similar trends using either K2P or the GTR distances, and as expected, at high levels of DNA differences, GTR model yields a comparatively higher DNA distances.
Appendix References
- Felsenstein, J. 1993. PHYLIP (Phylogeny Inference Package) version 3.5c, Seattle, WA.
- Kimura, M. 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16:111-120.
- Learn, G. H., B. T. M. Korber, B. Foley, B. H. Hahn, S. M. Wolinsky, and J. I. Mullins. 1996. Maintaining the integrity of HIV sequence databases. J. Virol. 70:5720-5730.
- Smith, S. W., R. Overbeek, C. R. Woese, W. Gilbert, and P. M. Gillevet. 1994. The Genetic Data Environment: An expandable GUI for multiple sequence analysis. CABIOS. 10:671-675.
- Swofford, D. L. 1999. PAUP 4.0: Phylogenetic Analysis Using Parsimony (And Other Methods), version 4.0. Sinauer Associates, Inc.